The genetics of cleft lip and palate
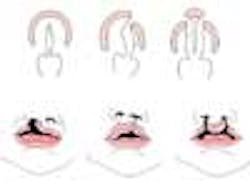
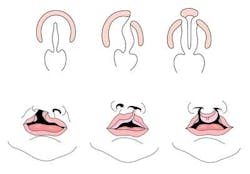
Cleft lip with or without cleft palate (CL/CP) differs from an isolated cleft palate (CP) on embryonic, epidemiologic, and genetic levels. Cleft lip typically results from the maxillary prominence and medial nasal prominence failing to fuse between the fifth and sixth week of embryonic development. Normal palate development results from the formation of the primary palate and secondary palate. The primary palate is formed at weeks six to seven by the development and fusion of the medial nasal, lateral nasal, and maxillary processes. The secondary palate originates from the palatal shelves (that develop from the paired maxillary processes of the first branchial arch) becoming horizontal and fusing, forming the hard and soft palates at around the ninth week of embryonic development. Shelves also fuse with the primary palate and nasal septum. (1)
Oral clefts are one of the most common birth defects seen in the neonatal nursery, with an overall prevalence of 1.6 per thousand newborns worldwide, with CL/CP seen in approximately one per thousand births and CP seen in 0.6 per thousand births. (2) There is a higher frequency of CL/CP in individuals of Asian, African, and Native American descent. CL/CP is also more common in males. In contrast, there is no significant difference in incidence of CP among different ethnic backgrounds, and CP is more common in females. (3) Risks for recurrence within a family depend on whether the cleft is isolated (with no other clinical findings present) or seen as part of a genetic syndrome. Most cases of oral clefts are isolated (approximately 80%). Isolated clefts are believed to have multifactorial inheritance: they’re due to a combination of multiple factors, both genetic and environmental. The risk for recurrence (Table 1) increases when there is more than one affected relative. The risk for recurrence also increases the more severe the defect is.

Cleft lip and palate can be seen with other congenital anomalies. The likelihood of a genetic or teratogenic etiology increases the more congenital anomalies with which a patient presents. Presence of other issues such as intellectual disability, behavioral problems such as autism, dysmorphic features, or other medical concerns will also make a genetic disorder or a teratogenic exposure more likely. Approximately 13% of individuals with cleft lip will have other medical concerns or anomalies. The number increases to 37% with cleft lip and palate and to 47% with cleft palate alone.
Prenatal exposure to teratogenic agents (such as thalidomide, anticonvulsants, alcohol, retinoic acid, and cigarettes) and maternal disease (such as diabetes, rubella, and folate deficiency) have been shown to increase the risk of oral clefts. The presence of amniotic bands also increases the risk of clefts. Periconceptual folic acid supplementation is known to reduce the risk of oral clefts.
Pierre Robin sequence is a craniofacial anomaly characterized by mandibular hypoplasia or micrognathia, secondary U-shaped cleft palate, and glossoptosis leading to obstructive apnea and feeding difficulties. Pierre Robin sequence can be seen as part of genetic syndromes (22q11.2 deletion syndrome, Stickler syndrome; described below). (5)
There are hundreds of genetic syndromes associated with oral clefts, including cytogenetic abnormalities (aneuploidies, microdeletions) and single-gene (Mendelian) disorders. Confirming a genetic diagnosis is essential to determine prognosis and establish a risk for recurrence.
Aneuploidies such as Trisomy 13 and 18 have a strong association with CL/CP. Trisomy 13 (AKA Patau syndrome) is associated with three copies of chromosome 13, or unbalanced Robertsonian translocations involving chromosome 13. Babies born with this condition typically die in the neonatal period. Clinical features include cleft lip and palate, growth retardation, severe central nervous malformations (including holoprosencephaly), microcephaly, micropthalmia, iris coloboma, absence of the eyes, malformed ears, polydactyly, clenched fists, rocker bottom feet, congenital heart defects and urogenital defects. Midline clefts (otherwise very rare) can be seen in trisomy 13 due to the risk for midline defects, including holoprosencephaly. Trisomy 18 (AKA Edwards syndrome) is typically due to three distinct copies of chromosome 18, and is associated with poor postnatal outcome. Clinical features include cleft lip and palate, intellectual disability, failure to thrive, congenital heart disease, hypertonia, micrognathia, short sternum, low set malformed ears, clenched hands, rocker bottom feet, and hypoplastic nails, among others. Trisomy 13 and 18 can be easily confirmed or ruled out by doing chromosome analysis (karyotyping).
Microdeletion syndromes typically involve the deletion of part of a chromosome. These deletions may be too small to be detected by standard karyotyping and may require FISH (fluorescence in situ hybridization) or microarray technology to be detected. A well-known microdeletion syndrome associated with cleft palate is 22q11.2 deletion syndrome (aka Digeorge/Velocardiofacial syndrome). Palatal abnormalities including velopharyngeal incompetence, submucosal clefts, bifid uvula, and cleft palate are seen in 69% of individuals with 22q11.2 deletion, and can be part of the Pierre Robin sequence. Other clinical findings include congenital heart disease, hearing loss, dysmorphic features, immune deficiency, hypocalcemia, renal anomalies, feeding issues, skeletal anomalies, and psychiatric disorders. Approximately 10% of cases of 22q11.2 deletion syndrome are believed to be familial. The deletion segregates in an autosomal dominant fashion.(6) Wolf-Hirschhorn syndrome, which is due to a deletion in the short arm of chromosome 4, is also associated with oral clefts (in 25% to 50% of affected individuals). Characteristic facial features (including prominent glabella leading to “Greek-warrior helmet appearance”), congenital heart disease, intellectual disability, seizures, failure to thrive, micrognathia, preauricular tags or pits, and hypodontia can also be seen as part of the condition.(7)
Single-gene disorders with oral clefts include Stickler syndrome, Treacher Collins syndrome, and Van der Woude syndrome, among many others. Stickler syndrome is a collagen disorder with autosomal dominant and, less commonly, autosomal recessive inheritance. Common features include cleft palate (seen as part of Pierre Robin sequence or without micrognathia), hearing loss (sensorineural and conductive), skeletal findings (early onset arthritis, spondyloepiphyseal dysplasia), ocular anomalies (high myopia, vitreous abnormalities) and characteristic facial features (with underdevelopment of the maxilla and nasal bridge, midface retrusion). Genetic testing for Stickler syndrome can be complex, as mutations in at least six genes have been described in affected individuals. Approximately 90% of patients with Stickler syndrome have mutations in the COL2A1 gene and have an autosomal dominant form of the condition.(8) Treacher Collins syndrome is an autosomal dominant condition characterized by cleft palate with or without cleft lip in 28% of affected individuals. Other abnormalities include hypoplasia of the zygomatic bones and mandible, external ear anomalies, coloboma of the lower eyelid, conductive hearing loss, absence of lower eyelashes, preauricular hair displacement onto the cheeks, and choanal stenosis or atresia. The diagnosis of Treacher Collins syndrome is based on clinical and radiographic findings. Mutations in at least three genes have been described, with mutations in TCOF1 seen in 78% to 93% of patients.(9) Van der Woude syndrome is characterized by the presence of congenital, usually bilateral, paramedian lower-lip fistulae (pits), or sometimes small mounds with a sinus tract leading from a mucous gland of the lip, and oral clefts (including CL/CP and CP). Van der Woude is an autosomal dominant condition associated with mutations in the IRF6 gene (10). Testing for single-gene or multi-gene conditions requires direct analysis of the gene by sequencing and/or deletion/duplication analysis (such as MLPA).
Given that genetic syndromes with cleft lip and palate can be associated with aneuploidies, chromosome microdeletions/microduplications, or single-gene disorders, genetic testing can be a complicated process. A thorough medical history, a three-generation pedigree, a pregnancy history, and a dysmorphology exam by a clinical geneticist may clarify the clinical picture and allow for targeted genetic testing. Newer technologies including microarray will allow for the identification of small microdeletions and microduplications previously missed by standard karyotyping. Unfortunately, this technique also leads to the identification of deletions and duplications of unknown clinical significance, complicating the genetic counseling process. Testing for single-gene disorders or Mendelian disorders requires the clinical availability of genetic testing for the desired gene. It can also be expensive if not covered by medical insurance. New technologies such as Next-Generation sequencing, exome sequencing, or genome sequencing (known collectively as genomic tests) have now become clinically available. By analyzing hundreds to thousands of genes simultaneously, these tests significantly increase diagnostic power and yield. Compared to other techniques, these tests can provide an answer faster and in a more cost-efficient manner. In the research field, exome and genome sequencing has led to the identification of new genes as well as expansion of the clinical features and spectrum for genetic mutations. As with microarray technology, genomic tests can detect syndromes that are unrelated to the patient’s presentation and/or reason for testing. Given the inherent complexities of genetic testing, informed consent is necessary.
Conclusion
Although cleft lip and palate is an isolated anomaly in the majority of cases, there is a strong association between oral clefts and other anomalies and genetic syndromes. A genetic evaluation by a clinical geneticist and a genetic counselor is essential for anticipatory guidance and to determine risks for recurrence. Genetic testing, which requires informed consent, can be coordinated and interpreted during a genetic evaluation.
Anya Revah, MS, is the senior genetic counselor at the Division of Medical Genetics at Maimonides Infants and Children's Hospital in Brooklyn, New York. She is also an active member of the Maimonides Medical Center and Kings County Hospital Cleft Lip and Palate Multidisciplinary Team. She has a Master's in Science in Genetic Counseling from Boston University in Boston, Massachusetts.
References
1. Sadler TW. Langman’s Medical Embryology. Ninth Edition. Pages 390-395.
2. Parker SE, Mai CT, Canfield MA, Rickard R, Wang Y, Meyer RE, Anderson P, Mason CA, Collins JS, Kirby RS, Correa A. For the National Birth Defects Prevention Network. Updated national birth prevalence estimates for selected birth defects in the United States. 2004-2006. Birth Defects Research (Part A): Clinical and Molecular Teratology 2010;88:1008-1016.
3. Fraser FC. The genetics of cleft lip and cleft palate. Am. J. Hum. Genet. 1970;22: 336–352.
4. Van Rooij IA, Ocke MC, et al. Periconceptual folate intake by supplement and food intake reduces the risk of non-syndromic cleft lip with or without cleft palate. Prev Med 2004;39: 689-694.
5. Tan TY. Kilpatrick N, Farlie PG. Developmental and genetic perspectives on Pierre Robin sequence. Am. J. Med. Genet. 2013;163C:295-305.
6. McDonald-McGinn DM, Emanuel BS, Zackai EH. 22q11.2 Deletion Syndrome. Sept. 23, 1999. [Updated Feb. 28, 2013]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1523/.
7. Battaglia A, Carey JC, South ST, et al. Wolf-Hirschhorn Syndrome. Apr. 29, 2002. [Updated 2010 Jun 17]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1183/.
8. Robin NH, Moran RT, Ala-Kokko L. Stickler Syndrome. Jun. 9, 2000. [Updated Sept. 11, 2014]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1302/.
9. Katsanis SH, Jabs EW. Treacher Collins Syndrome. Jul. 20, 2004. [Updated Aug. 30, 2012]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1532/.
10. Schutte BC, Saal HM, Goudy S, et al. IRF6-Related Disorders. Oct. 30, 2003. [Updated Jul 3, 2014]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2014. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1407/.